What is GenoML?
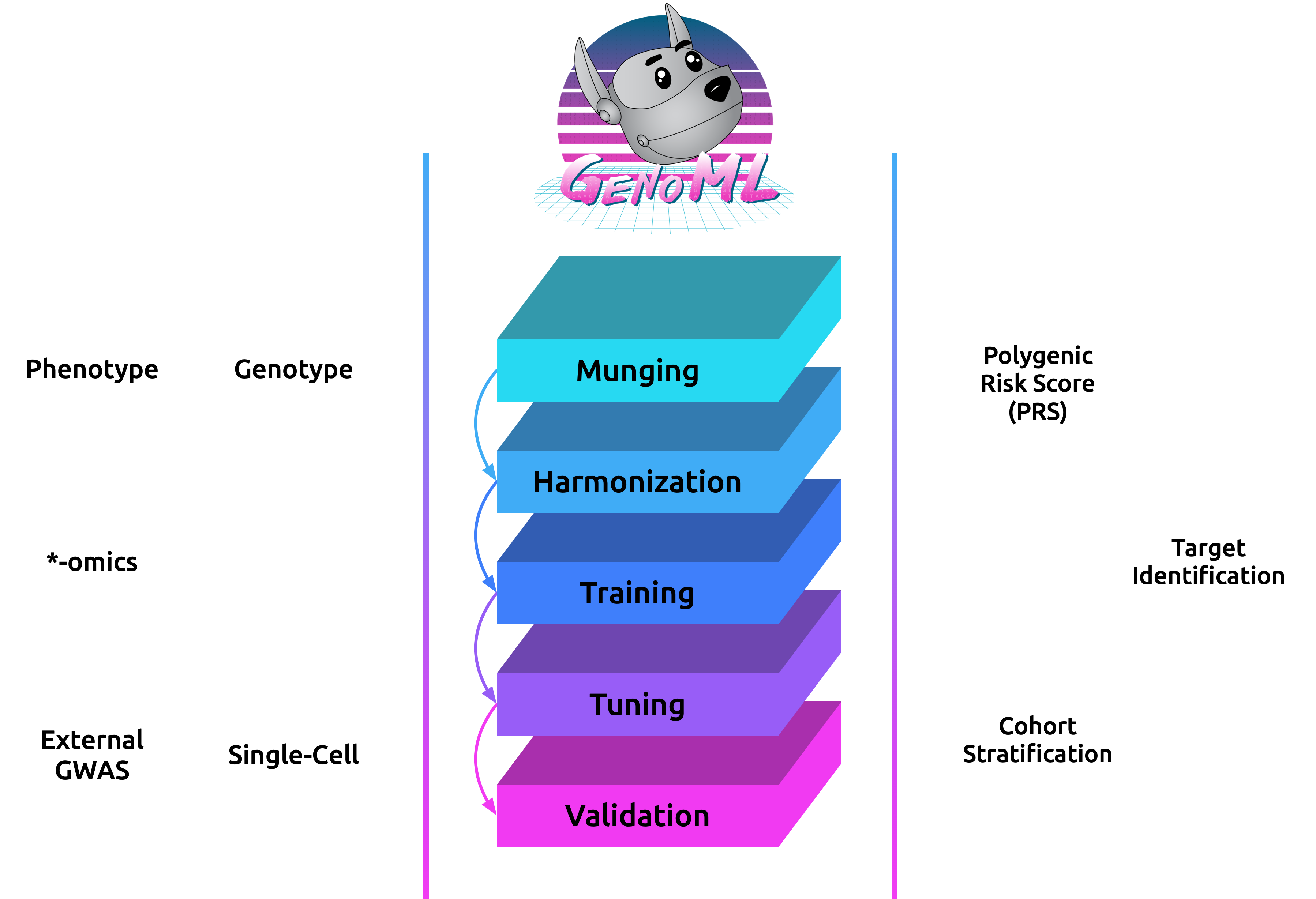
GenoML is a Python package automating machine learning workflows for genomics (genetics and multi-omics) with an open science philosophy. Genomics data require significant domain expertise to clean, pre-process, harmonize and perform quality control of the data. Furthermore, tuning, validation, and interpretation involve taking into account the biology and the limitations of the underlying data collection, protocols, and technology. GenoML’s mission is to bring machine learning for genomics and clinical data to non-experts by developing an easy-to-use tool that automates the full development, evaluation, and deployment process. Emphasis is put on open science to make workflows easily accessible, replicable, and transferable within the scientific community.
Backgound#
In recent years, the demand for machine learning (ML) expertise has outpaced the supply, despite the surge of people entering the field. To address this gap, there have been significant strides in the development of user-friendly machine learning software that can be used by non-experts. The first steps toward simplifying machine learning involved developing simple, unified interfaces to a variety of machine learning algorithms (e.g., scikit-learn, XGBoost, LightGBM, TensorFlow, PyTorch). Although these packages have made it easy to experiment with machine learning, there is still a fair bit of knowledge and background in data science required to produce high-performing and usable machine learning models. This demand has given rise to the area of automated machine learning (AutoML). Some of the recently developed and popular AutoML systems include Auto-WEKA, hyperopt-sklearn, Auto-sklearn, TPOT, and Auto-Keras.
However, different data require different ML pipelines. The development of ML models for genomics (genetics and multi-omics) data, in particular, is notoriously difficult for a non-expert. These data modalities require significant domain expertise to clean, pre-process, harmonize and perform quality control (QC). Furthermore, tuning, validation, and interpretation involve taking into account the biology and the limitations of the underlying data collection, protocols, and technology.
GenoML as a package#
For ML to truly be accessible to non-experts in the genomics and clinical research areas, we have designed an easy-to-use tool, called GenoML, that automates the full development, evaluation, and deployment process. GenoML provides an end-to-end framework for genomic datasets, including the most complex parts of the process, such as data pre-processing and cleaning, to more advanced training and tuning. GenoML intelligently explores many possible techniques to find the best model for the specific input data. GenoML is also helpful to advanced users; it provides a high-level wrapper performing many modeling tasks that would typically require many more lines of code.
GenoML as a community#
GenoML is more than a package. Since its inception, it has evolved into a diverse community with integrative expertise in data science, bioinformatics, computer science, software engineering, biology, and healthcare. GenoML contributors are staunch advocates of #OpenScience, striving to make data and code easily accessible to the scientific community. Please join us and contribute to the development of GenoML.
Issues, Suggestions, or Need Help?#
If anything is broken, confusing, or you just want new features, please hit us up on Twitter or submit a ticket to our GitHub issues.
If you want to try and troubleshoot your issue by yourself, please try running your command with the "-v "or "-vvv "options at the end to generate verbose outputs. If you'd like to get more involved in GenoML and the community we are trying to build, please see the collaborations page for additional info.
Team#
Core#
Mary Makarious (NIA-NIH / UCL) Hampton Leonard (NIA-NIH / DTI / DZNE) Dan Vitale (NIA-NIH / DTI) Sayed Hadi Hashemi (UIUC) David Saffo (NEU) Eduardo Salmerón Castaño (UM) Cornelis Blauwendraat (NIA-NIH) Hirotaka Iwaki (NIA-NIH / MJFF / DTI) Roy H. Campbell (UIUC) Andrew B. Singleton (NIA-NIH) Juan A. Botia (UM) Faraz Faghri (NIA-NIH / DTI) Mike A. Nalls (NIA-NIH / DTI)
Collaborators#
Lana Sargent (VCU) Susan Chacko (Biowulf-NIH) Rafael Jordá Muñoz (UM)
Affiliations#
- Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health (LNG-NIA-NIH)
- Center for Alzheimer's and Related Dementias, National Institutes of Health (CARD-NIH)
- Data Tecnica International (DTI)
- Systems Research Group, Department of Computer Science, University of Illinois at Urbana-Champaign (UIUC)
- UCL Movement Disorders Centre, University College London (UCL)
- German Center for Neurodegenerative Diseases (DZNE)
- College of Computer Sciences, Northeastern University (NEU)
- The Michael J. Fox Foundation (MJFF)
- Biowulf High-Performance Computing Cluster, National Institutes of Health (Biowulf-NIH)
- School of Nursing, VCU Health, Geriatric Health Clinic, Virginia Commonwealth University (VCU)
- University of Murcia (UM)
Citations#
If using GenoML for a publication, we ask that you cite following paper:
- Makarious, M. B., Leonard, H. L., Vitale, D., Iwaki, H., Saffo, D., Sargent, L., Dadu, A., Castaño, E. S., Carter, J. F., Maleknia, M., Botia, J. A., Blauwendraat, C., Campbell, R. H., Hashemi, S. H., Singleton, A. B., Nalls, M. A., & Faghri, F. (2021). GenoML: Automated machine learning for genomics. ArXiv:2103.03221 [Cs, q-Bio]. http://arxiv.org/abs/2103.03221
Acknowledgement#
This work is a collaborative effort in open source software supported by the Laboratory of Neurogenetics, and the Center for Alzheimer's and Related Dementias at the National Institute on Aging (National Institutes of Health), the Biowulf High-Performance Computing Cluster (National Institutes of Health), the Michael J Fox Foundation, University of Murcia, the Global Parkinson's Genetics Program, Chan Zuckerberg Initiative, and Data Tecnica Int'l.
GenoML would not be possible without these excellent resources from the python and scientific community (let's give some credit where it's due): joblib, pyparsing, numpy, kiwisolver, six, cycler, python-dateutil, matplotlib, numexpr, tables, pytz, pandas, pyyaml, toolz, locket, partd, fsspec, dask, packaging, zipp, importlib-metadata, pluggy, wcwidth, more-itertools, py, attrs, pytest, pycparser, cffi, zstandard, xarray, tqdm, wrapt, pandas-plink, plink, urllib3, idna, chardet, requests, threadpoolctl, scipy, scikit-learn, seaborn, patsy, statsmodels, xgboost.